|
Sophia Yancopoulos, Mihaly Mezei, Roman Osman
Originally presented as poster
210 February, 2001 at Annual Biophysical Society Meeting in
Boston, MA |

|
|
Sophia Yancopoulos, Mihaly Mezei, Roman Osman
Originally presented as poster
210 February, 2001 at Annual Biophysical Society Meeting in
Boston, MA |
|
The recent crystallographic structures of the
D96N mutant of bacteriorhodopsin
(Luecke et al, 1999)in
the ground (BR) and the excited (M) states reveal internal waters with
proposed specific functionality. We have investigated the localization
of the internal waters with grand canonical ensemble Monte Carlo
simulations.
The most probable positions of simulation sites were determined with the
generic site method. The results of the simulations show 3 clusters of
internal sites in each state and additional isolated sites in hydrophobic
pockets. Ten of the 11 internal BR state waters are well matched by the
simulations at an occupancy-weighted average distance of 0.69 ±
0.5 A. Three additional internal sites with total occupancy of 1.71 have
been found for the BR state and 11 additional in the M state with total
occupancy of 6.4 waters. Superimposing the two data sets yields 14 water
pairs with an average distance of 0.82 ± 0.45 A. The simulation
locates only one of three missing waters near Asn-96 in the M
state.Supported
by Training Grant DK07645
INTRODUCTION
Water is an in integral component
of proteins and plays an important role in their structure and function.
High resolution structures determined by x-ray crystallography have recently
become available for bacteriorhodopsin, a 7 TM protein, and its mutants
(Luecke
et al, 1999). The waters in these structures have been clearly observed
and offer an excellent opportunity to test theoretical methods of solvation.
Furthermore, simulations offer an opportunity to investigate the properties
of internal waters and their role.
METHODS
In
order to explore the placement of water in the transmembrane domain of
the 7 TM protein bacteriorhodopsin we have used a Grand Canonical Ensemble
Metropolis Monte Carlo (MMC) cavity biased insertion method (Resat and
Mezei, 1994). This method has already been successful in determining water
positions in a number of crystal structures, for example, small
polydisaccharides
(Resat & Mezei, 1994), DNA (Resat & Mezei, 1996), and
complexes of inhibitors bound to HIV-1 protease (Marrone
et al.1998). The cavity-biased formulation of MMC was chosen since
it improves statistical sampling efficiency (Mezei 1987) and is therefore
an appropriate method to investigate the solvation of the TM domain of
BR.
Cavity
Biased GRAND CANONICAL ENSEMBLE MMC
In this approach the insertion of
a molecule is attempted if a cavity of appropriate radius is found and
accepted with a probability:
P' = min { 1,Pcav(N) exp[-(B+E(N+1)- E(N))/kT+B] / (N+1)}
where E(N) is the potential energy of the system of N particles and Pcav(N) is the probability of finding a cavity of a specific size.
The parameter B is related to the excess chemical potential as
µ' = kT (B - ln <N>)
Where <N> is the average over the number of
particles.
TUNING THE B
PARAMETER
The scheme is implemented by first adjusting the B parameter until the targeted number of molecules of solvent is approximately achieved outside a rectangular region containing the molecule so that the density in this region external to the protein approaches the density of water (0.997 grams per cc). Water molecules are then inserted or deleted by accepting or rejecting each attempt based on the Metropolis Monte Carlo criterion.
The tuned values of the B parameter
were respectively: -1.2 and ?1.35 for the BR and M states.
CALCULATION:
The atomic coordinates of the BR (unphotolyzed) state, observed at 1.8 Angstrom resolution, and M (excited) state observed with a resolution of 2Å, from the Protein Data Bank (accession numbers 1C8RR and 1C8S respectively). The waters and lipids were removed from the structures. Hydrogens were added to the structures with CHARMM. The N-and C-termini as well as two residues, which were discontinuous in the original structures, were capped with acetyl and N-methyl groups, respectively.
The BR and M states were run
for
5 million and 6 million MMC steps, respectively, with the equilibrated
B parameter. The coordinates of the atoms in the systems were stored every
10,000 steps during sampling. During the simulations the solute
residues
were kept fixed at their experimental crystal structures and only the water
molecules were allowed to move. This was in keeping with prior MMC
simulations
(Resat and Mezei 1996). Periodic boundary conditions were applied with
the cell size as determined by simulaidês OPT. The temperature was
298 K. The water model chosen was the TIP3P (Jorgensen et al., 1983) with
the solute potential type calculated in CHARMM. The solute-solvent
cutoff was used with the minimum-image convention. The solvent spherical
cutoff was 10 Å. The resultant average densities obtained in
the exterior region were 0.99875 g/ml and 0.99743 g/ml
respectively
in the BR and M states.
LOCATING
GENERIC SITES:
To establish the positions of water
sites from the simulation, we utilized the generic solvent (GS) site
method developed by Mezei and Beveridge, 1984. The configurations
from the trajectory are superimposed and the generic sites are determined
by the graph theoretical method, (Berge, 1962), which minimizes the
root mean square deviation between the GS and the individual waters assigned
to it in each snapshot. The GS are characterized by mean
locations
for the oxygen positions, the mean square deviation of the position, and
the occupancy of the site: the ratio of the number of water molecules
assigned
to the location divided by the total number of configurations.
BR STATE RESULTS:
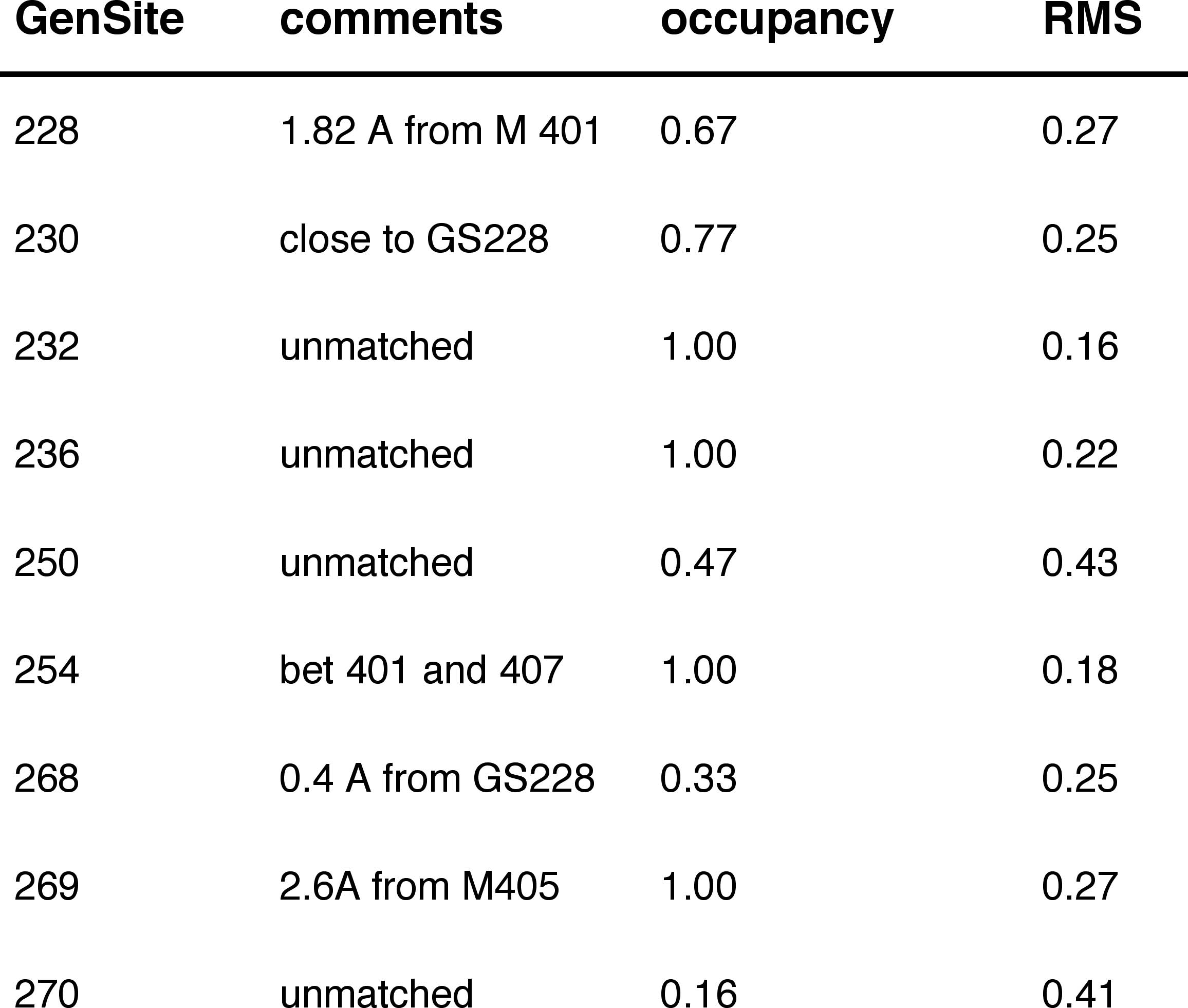
Table 1a: Internal BR State Generic Sites matching Crystallographic Waters
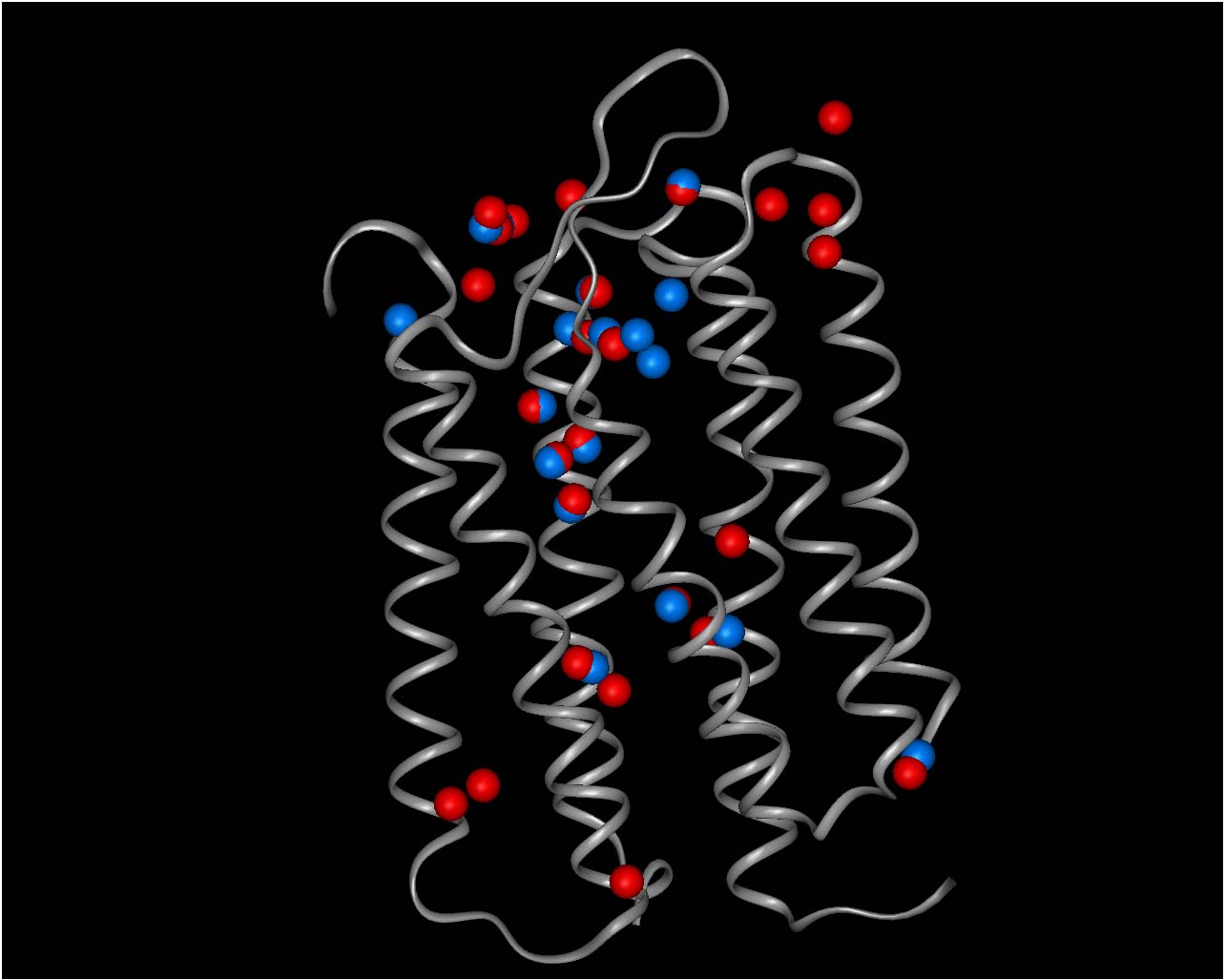
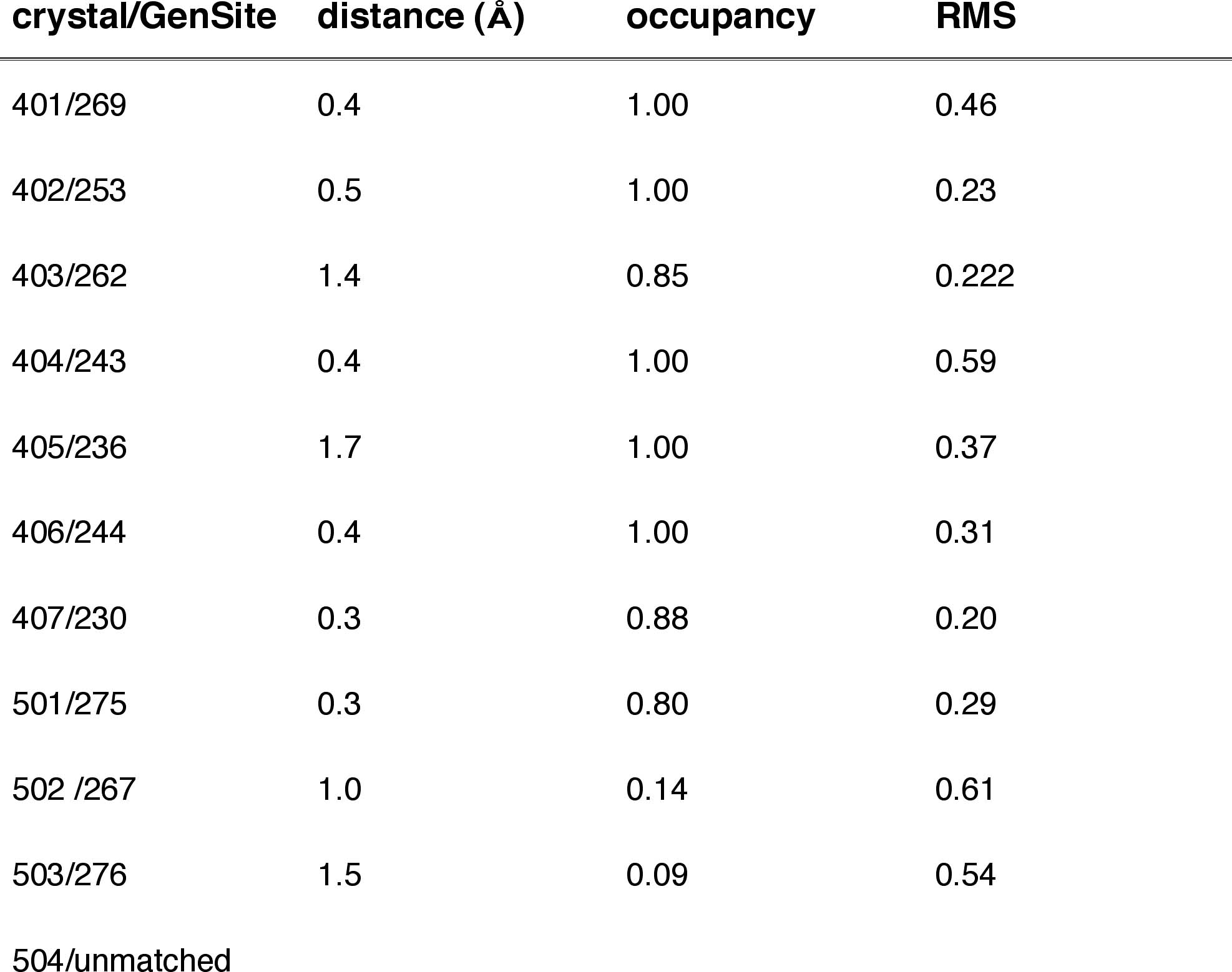
Figure 1.(blue)
BR state simulation waters superimposed on
(red) X-ray positions.
EXPLANATION FOR MISSING
AND LOW OCCUPANCY WATERS:
Two factors influence the occupancy
of a site. One is the potential energy of the water at a particular
insertion
site. The other is the B factor, which controls the equilibration between
the excess chemical potential and the average number of particles in the
GC ensemble.
Energies of Representative Sites in k-cal/mol
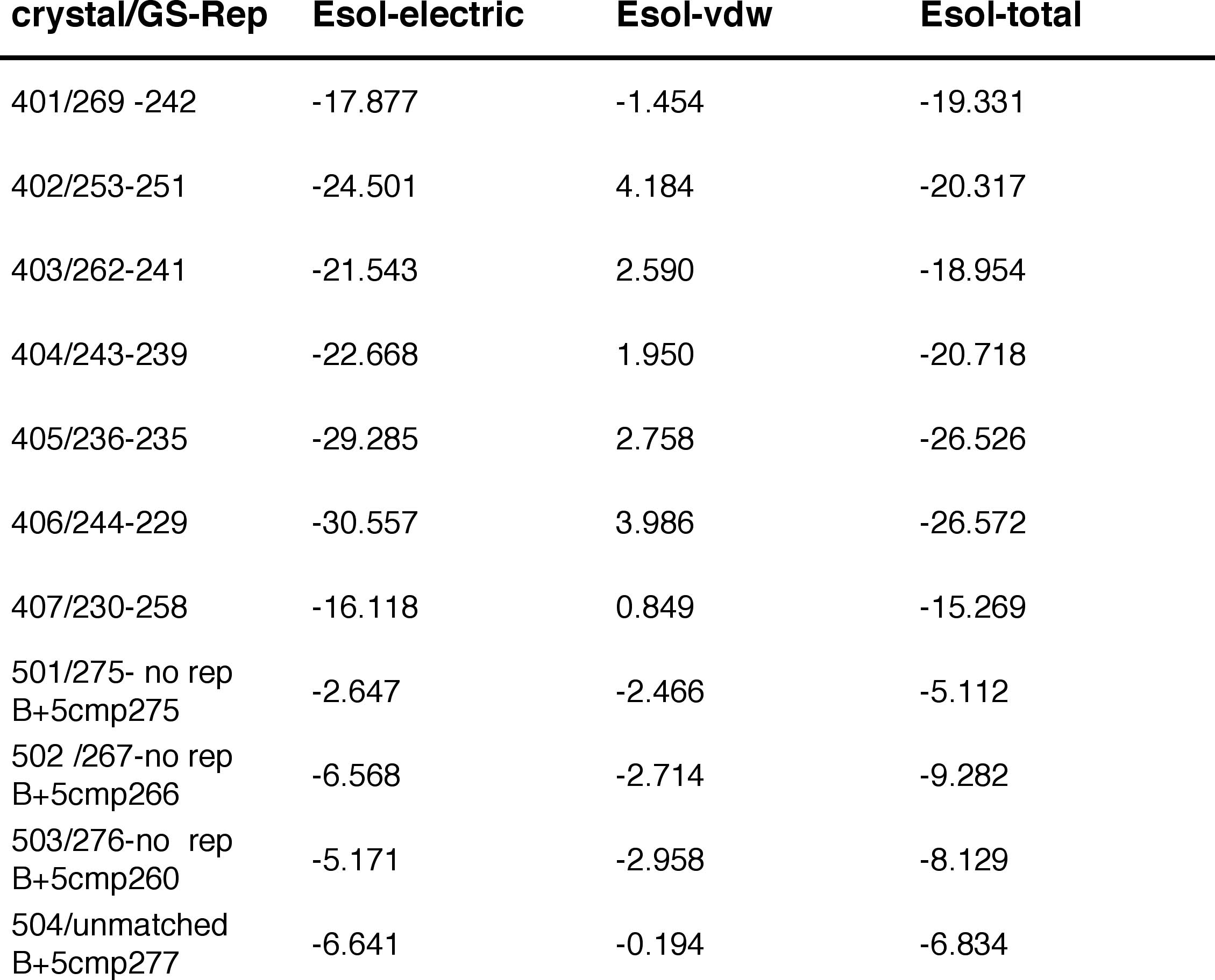
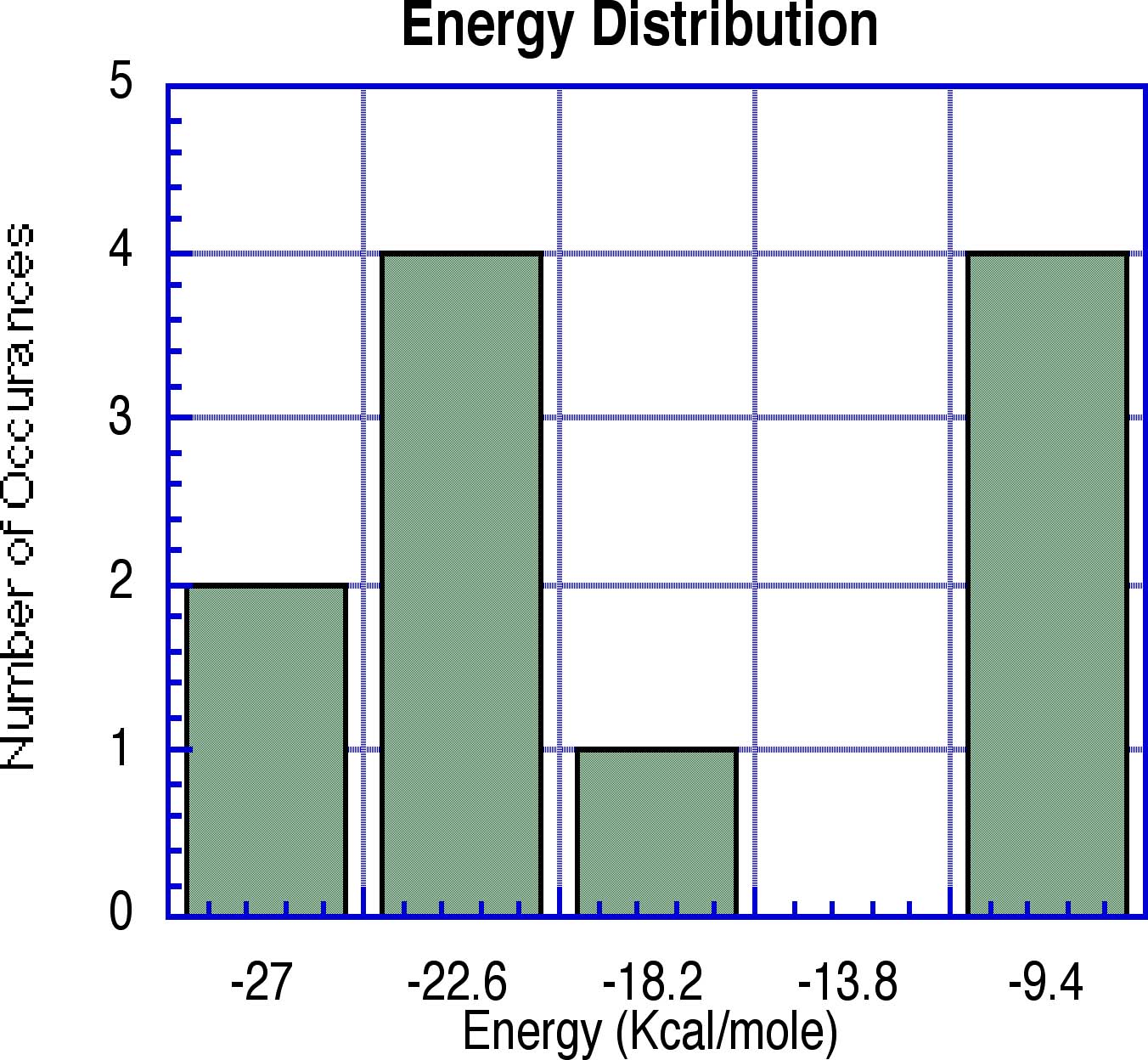
Conclusion: Waters in low
occupancy sites have significantly higher energies.
Occupancy as a function of B:

Increasing the excess chemical
potential (i.e., increasing B) progressively increases the occupancy
by overcoming the unfavorable energy.
Simulations find Additional
Waters:
Table 1b: Internal BR Generic Sites withoutmatching Crystallographic Waters:
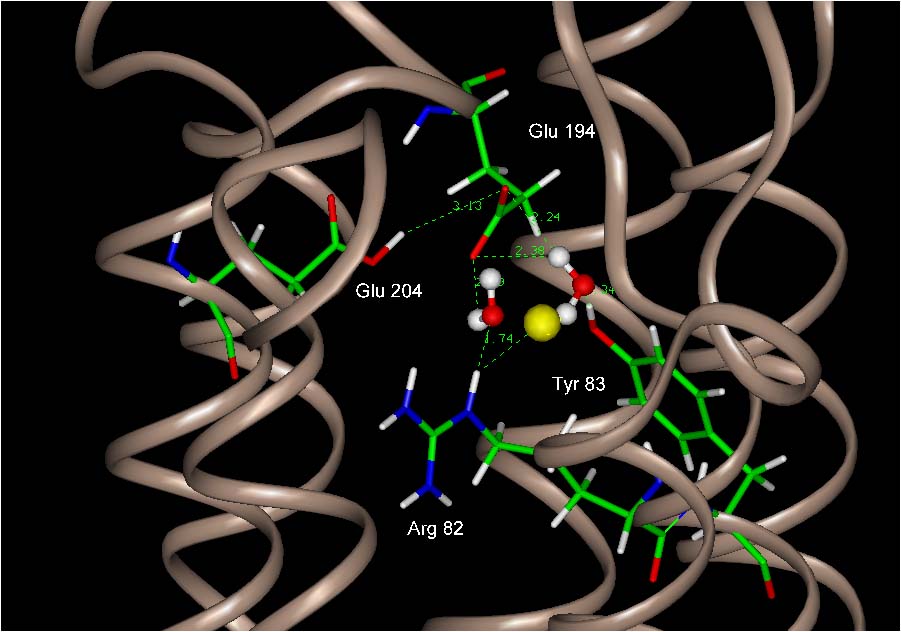
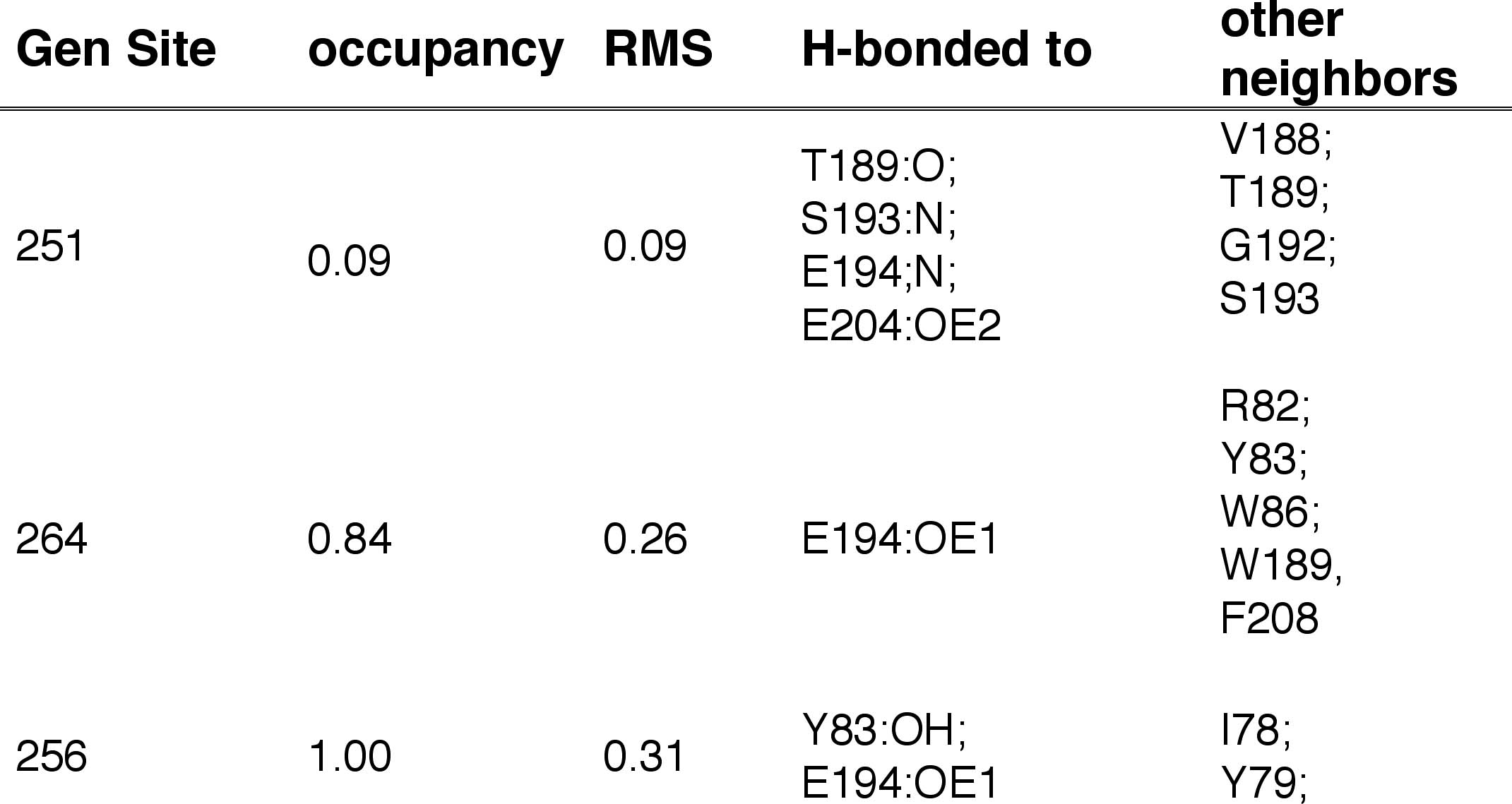
FIGURE 2: A
snapshot from the simulation. BR state Crystallographic water 405 is
shown in yellow. One match Simulation water 236 is to the left. An
additional match, site 256 is on the right . Another two
nearby simulation waters, 251 and 264, are not seen in this view,
but their properties are summarized in the table and
below.
PROPERTIES OF ADDITIONAL WATER
SITES :
CONCLUSIONS:
Additional waters are placed in plausible positions.
MMC is successful at locating
potentially real water sites that were not observed
crystallographically.
M STATE RESULTS
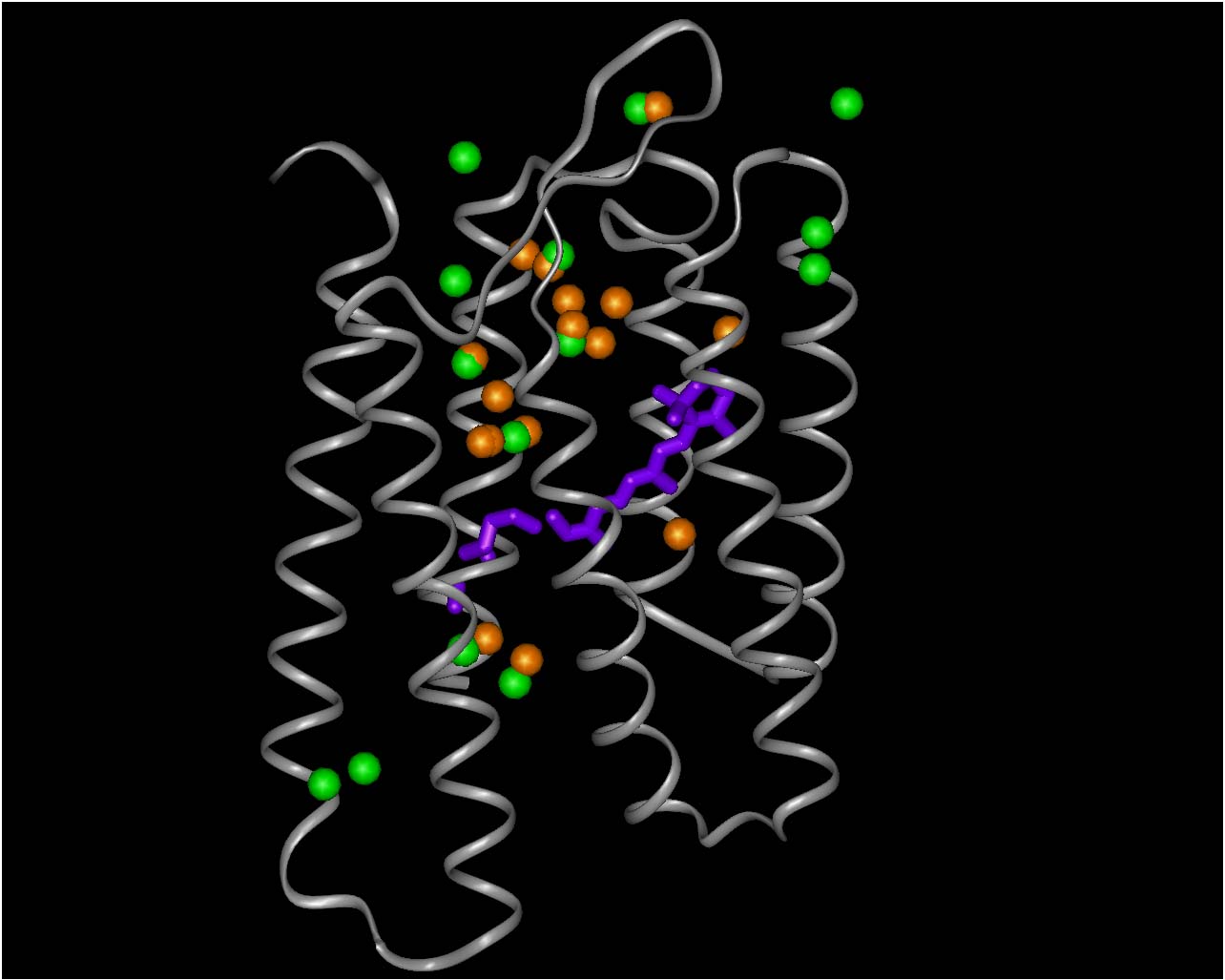
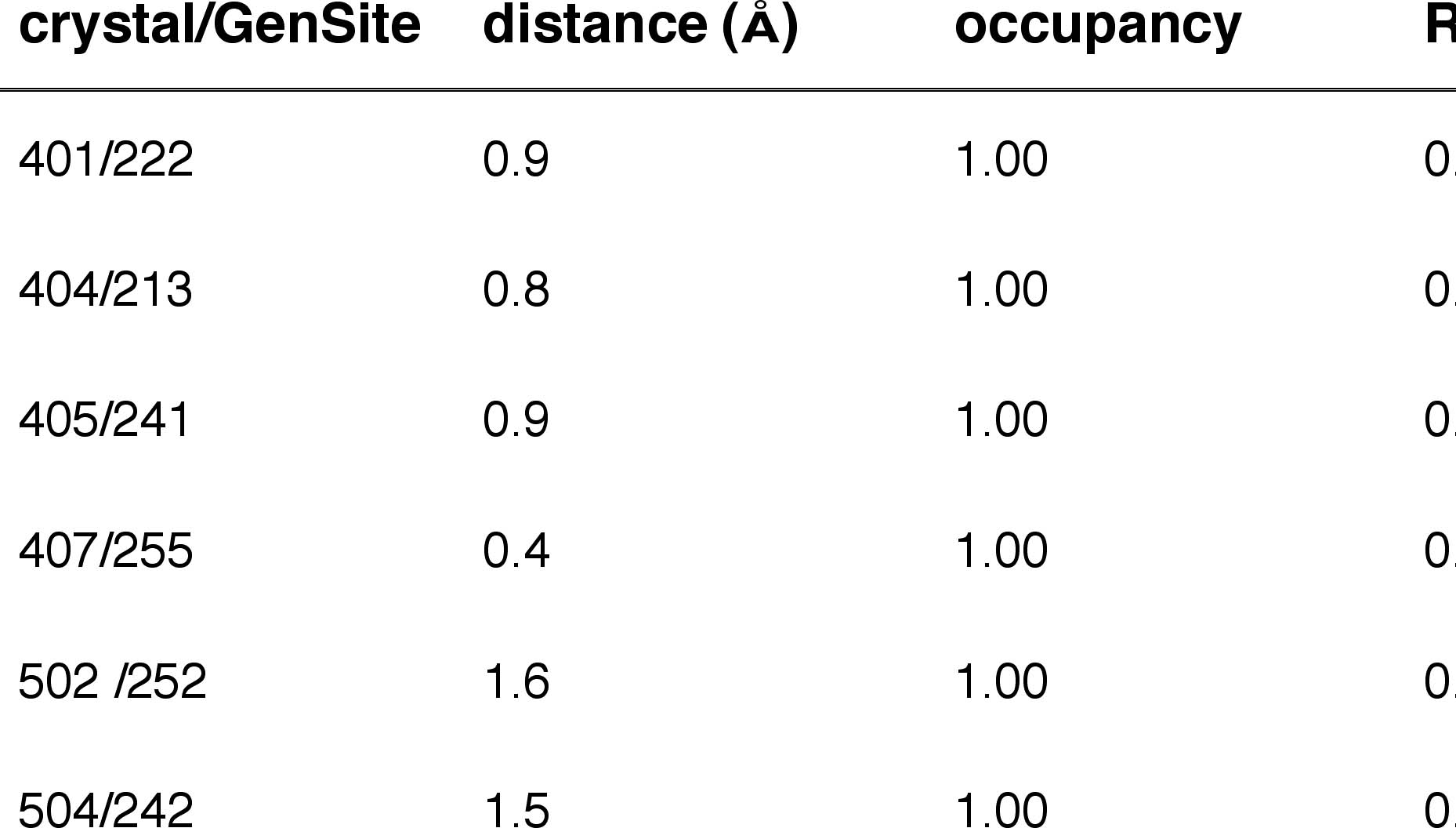
FIGURE 3: M state crystallographic waters
in orange, simulation waters in green
OBSERVED M CRYSTALLOGRAPHIC
WATERS ARE MATCHED WELL BY SIMULATIONS AND HAVE HIGH OCCUPANCY
.
ADDITIONAL M STATE SITES ARE
FOUND WHICH DO NOT MATCH M STATE X-RAY WATERS:
|
CONCLUSIONS:
1. The Grand Canonical cavity-biased Monte Carlo Method is a reliable approach for internal water localization.
2. Additional waters placed by the method in the BR state reside in an H-bonded network environment.
3. M state has the same number of internal waters as BR state.
4. In M state, new locations
of water sites are identified, suggesting that they were formed by the
conformational change in bacteriorhodopsin.
REFERENCES
D.J. Adams, Grand canonical ensemble Monte Carlo
for a Lennard-Jones fluid, Molec. Phys., 29, 311 (1975).
H. Luecke, B. Schobert, H. T. Richter, J. P. Cartailler, and Janos K. Lanyi, Structural Changes in Bacteriorhodopsin During Ion Transport at 2 Angstrom Resolution Science,286, No 5438, 255-260 (1999)
T.J. Marrone, H. Resat, C.N. Hodge, C.-H. Chang, and J.A. McCammon, Solvation studies of DMP323 and A76928 bound to HIV protease: Analysis of water sites using grand canonical Monte Carlo simulations, Protein Science, 7, 573-579 (1998).
N.A. Metropolis, A.W. Rosenbluth, M.N. Rosenbluth, A.H. Teller, and E. Teller, Equation of state calculation by fast computing machines, J. Chem. Phys., 21, 1087 (1953).
M. Mezei, A Cavity-Biased (T,V,mu) Monte Carlo Method for the Computer Simulation of Fluids, Molec. Phys., 40, 901-906 (1980);
M. Mezei, Grand-canonical ensemble Monte Carlo study of dense liquids: Lennard-Jones, soft spheres and water. Molec. Phys., 61, 565 (1987); erratum: 57, 1207 (1989).
M. Mezei, On the Selection of the Particle to be Perturbed in the Monte Carlo Method, J. Comp. Phys., 39, 128-136 (1981).
M. Mezei, K.A. Bencsath, S. Goldman, and S. Singh,
The detailed balance energy-scaled displacement Monte Carlo algorithm,
Molecular
Simulation1, 87 (1987).
H. Resat, and M. Mezei, Grand Canonical Monte Carlo Simulation of Water Positions in Crystal Hydrates. J. Am. Chem. Soc.,116, 7451-7452 (1994).
H. Resat, and M. Mezei, Grand Canonical Ensemble Monte Carlo Simulation
of the dCpG/Proflavine Crystal Hydrate.
Biophys.
J.
71, 1179-1190 (1996).