Protocol for obtaining solvent sites with Grand-Canonical Ensemble simulations using MMC
Mihaly Mezei
Department of Phatmacological Sciences
Icahn School of Medicine at Mount Sinai
New York, NY 10019
Obtaining solvent sites requires the following steps when using MMC:
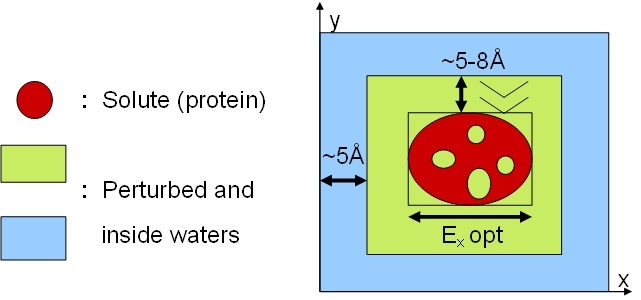
- Use Simulaid to optimize the orientation of your solute in a bounding box
(Note: NOT in a periodic cell!). Let it center the sysytem to (0,0,0) and
note the optimized edge lengths from Simulaid.s output:
=== Edges of the smallest box still enclosing the solute:
=== Optimum: 33.37131 48.39131 39.04523 A vol= 63053.4 A^3
Ex(opt) Ey(opt) Ez(opt)
- Use Simulaid to generate an .slt file (the solute description with potential codes).
This is one of the conversion options.
You may have to change PDB atom/residue names to Charmm (or Amber) convention first
(a other conversion option of Simulaid).
Unless you use a united atom model, make sure your structure contains the hydrogens too.
- Set up the GCE input for MMC (following the examples):
- Run a first a million MC steps with the B parameter set at the input
(with the GCEN key) to a positive number, say, 1.0.
This will mostly fill the simulation box.
- Repeatedly run 20 million (or more) steps until the number of waters stops increasing and the energy stops decreasing.
- Once the density is about to be settled to an acceptable value,
save every, say, 10000-th configuration in an .hst file (see the TRAJ and RUNS keys).
- Since this trajectory still contains lots of solvent waters,
use the FILT key to eliminate the outside waters.
The option we like uses the circular variance - a single cutoff can be chosen
to include waters only way inside, or near the surface, etc.
Alternative filtering can be done by solvation shells and solvation energies
(this latter is useful to get rid of waters solvating the membrane-bound side
of the protein.
- This filtered trajectory can be used to generate the so-called generic sites,
using the GENS key.
These sites will have RMS and fractional occupancy attached to them
so you will have some sense how certain MMC is in each.
- From the generic sites MMC will also create a representative configuration
(the configuration with low RMS from the sites found and few sites missing).
Also, MMC will scan the trajectory and for each site with occupancy
above a threshold value (specified on input) it will search for a solvent molecule
that is the closest to that site.
This way you will have a composite file with actual waters
in a reasonable orientation.
However, these waters may be inconsistent with each other (e.g., too close).
Finally, sites within a threshold distance can be clustered together
(these usually represent alternative sites with low occupancy each).
Last modified: 06/04/2019 (MM)